News
Research Unveils a Developmental Splicing Program Controlling Neuronal Maturation and Excitability
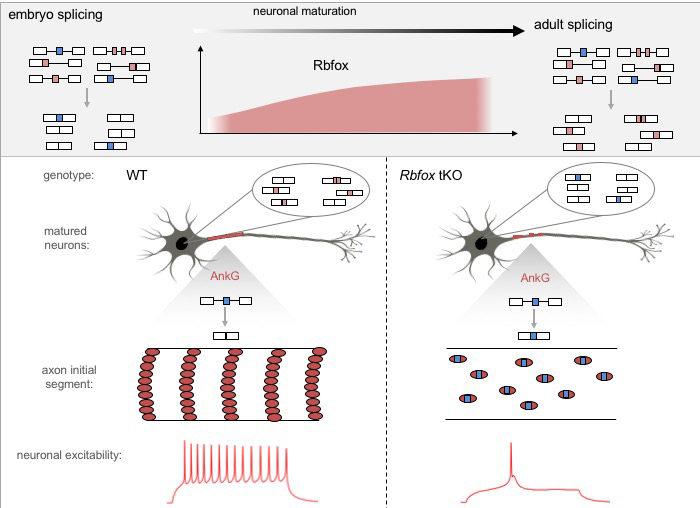
The Rbfox protein is a master regulator of neuronal RNA splicing that is demonstrated to be pivotal to establish the mature RNA splicing program in neurons. In neurons lacking this protein, an axonal cytoskeletal structure called “axon initial segment” is disrupted, impairing neuron’s ability to fire action potentials. (Image courtesy of Zhang Lab).
Neurons, or nerve cells, in the brain communicate with each other by transmitting electric signals, or firing action potentials, through long processes named axons (which send out signals) and dendrites (which receive signals). The capability of firing action potentials, among other functions of mature neurons, has to be acquired during development and neuronal maturation. However, the molecular mechanisms governing this complex process are so far poorly understood.
To peel away at the intricate layers that govern the development of neurons, a research team led by Chaolin Zhang, PhD, Assistant Professor in Systems Biology and Biochemistry and Molecular Biophysics and Hynek Wichterle, PhD, Associate Professor in Pathology & Cell Biology, Neuroscience, and Neurology , at the Center for Motor Neuron Biology and Disease , Columbia University Medical Center, focuses on a level of molecular regulation called alternative splicing. Alternative splicing is a process of generating multiple transcripts and protein variants by joining different combinations of coding segments. This process is highly dynamic during neural development with dramatic switches of splicing patterns in thousands of genes, which produce a repertoire of protein products required at specific developmental stages.
A key regulator of alternative splicing is the Rbfox family of proteins, which are enriched in neurons and previously were linked to neurodevelopmental disorders, including autism, schizophrenia, and epilepsy.
In a new study , published Feb. 1 in Neuron , Zhang, Wichterle, and their team determined that loss of Rbfox genes results in an “embryonic like” splicing program. Importantly, they observed a significant disruption of the assembly of the axon initial segment (AIS), a subcellular structure in the proximal part of the axon important for clustering of ion channels, and thus, for neurons to fire action potentials. Healthy neurons need action potentials to communicate with other cells. To this end, the team identified that Rbfox controls the splicing of the Ankyrin-G gene, which encodes the so-called “interaction hub protein” in the AIS, and found that the change of splicing in a small segment in this gene renders it unable to perform its normal function in organizing the AIS. In collaboration with Dr. Ke Xu’s lab at UC Berkeley, the team determined that this is because of aberrant accumulation and distribution of Ankyrin-G in the AIS.
“It is already known that Rbfox is important for controlling RNA splicing but what is not understood is its contribution to neuronal maturation and which specific cellular phenotypes it controls,” says Dr. Zhang. “We’ve singled out one important phenotype it controls, which leads to how neuronal excitability is controlled. Neurons communicate by firing action potentials, and if this process is disrupted then the neuron cannot do its job properly.”
Action potentials, also known as nerve impulses, are electrical messages sent from our brains to other organs and muscles; for instance, nerves in the brain transmit action potentials through neural networks to direct our arm muscles to contract in order to call into action our hands to lift or hold an item, a primary research focus at the Columbia Motor Neuron Center, to which both the Zhang and Wichterle labs are affiliated. Dr. Wichterle notes that “beyond their role in AIS assembly, our study identified a number of synaptic, cytoskeletal, and membrane proteins containing Rbfox regulated alternative exons. Rbfox proteins are dynamically regulated in response to neuronal activity and injury, raising the interesting possibility that Rbfox factors play a broader role in the regulation of neuronal plasticity and repair in the adult nervous system.”
In mammals, Rbfox proteins are made from three separate genes which are redundant with each other. Due to this redundancy, the study of Rbfox proteins in vivo is extremely challenging. To bypass the challenge, the team relied on their complementary expertise in genome engineering, stem cell differentiation into neurons, and genomic analysis of alternative splicing. The collaboration yielded the ultimate reference for Rbfox regulated exons and revealed their critical functional role in the control of neuronal maturation.
“This study is a great example of a highly collaborative endeavor that combines expertise in systems RNA biology, stem cell and neuronal biology, physiology, and cutting-edge imaging techniques. In fact, the first author, Martin Jacko, is jointly trained in the Wichterle lab and my lab, and I believe it is impossible to get to the point we are today without such intense collaboration,” said Zhang.
The researchers’ findings contribute to the mechanistic understanding of how the machinery required to initiate and propagate action potentials of neurons are controlled at the level of alternative splicing—a very tiny piece of the neuronal maturation puzzle, but one that has a significant impact. The study, titled “Rbfox Splicing Factors Promote Neuronal Maturation and Axon Initial Segment Assembly,” was published Feb. 1, ahead of print in Neuron .
The full list of authors: Martin Jacko, Sebastien M. Weyn-Vanhentenryck, John W. Smerdon, Rui Yan, Huijuan Feng, Damian J. Williams, Joy Pai, Ke Xu, Hynek Wichterle, and Chaolin Zhang.
The study was supported by grants from the National Institutes of Health, the Simons Foundation Autism Research Initiative, Project ALS Foundation, the Bakala Foundation, the China Scholarship Council, the Beckman Young Investigator Program, and the Packard Fellowships for Science and Engineering.